Sequence analysis of antifungal peptide
Introduction
A small cysteine-rich protein with antimicrobial activity was isolated
from pokeweed (Phytolacca americana) seeds and purified to homogeneity.
The protein inhibits the growth of several filamentous fungi and gram-positive
bacteria. The predicted three-dimensional structute shows a cystine-knot which is typical
in spider and other toxins. A positive patch and a hydrophobic patch of the model may provide
some hints for further wet-lab study.
Exercises
- Read the related papers published by Liu YF at ec.
Several NMR structures of anti-fungal peptide were reported in recent years.
Their coordinates were deposited to the Brookhaven Protein Data Bank (PDB).
The anti-fungal peptide (PDB code 1AFP) from Aspergillus giganteus consists
of 51 amino acid residues among them are 12 Lysines. The activity of this
peptide is the inhibition of the growth of a variety of filamentous fungi.
It has no effect on the growth of mammalian cells, yeast and eubacteria.
The structural feature of this peptide is a compact beta-barrel formed by
five anti-parallel beta-strands and stabilized by four internal disulfide
bridges [Campos-Olivas et al., 1995]. Interestingly, another anti-fungal
peptide isolated from radish (Raphanus sativus L.) seeds (1AYJ) poses the
same sequence length and same number of disulfide bridges as 1AFP. However,
no sequence similarity exists between these two peptides and the folding
unit of this peptide is not a beta-barrel, but a cysteine stabilized alpha-beta
motif [Fant et al., 1998]. This motif of three anti-parallel beta-strands
and an alpha-helix connected by three disulfide bridges has been found in
the scorpion toxin family. The biological activity of 1AYJ reduces elongation
of fungal hyphae and increases hyphal branching. A third peptide Drosomycin
induced by the fruit fly Drosophila melanogaster exhibits a potent activity
against filamentous fungi, but inactive against bacteria. This peptide is
7 residue shorter than the above two. Yet, the number of disulfide bridges
remains the same. The NMR structure of this peptide (1MYN) reveals that
it also belongs to the cysteine stabilized alpha-beta motif [Landon et al.,
1997]. The anti-fungal peptide we reported seems not belong to the above
two folding categories due to the small size in sequence length and the
number of the disulfide bridges.
Materials and Methods
Table 1 shows the structure templates used in the modeling
work. Seven templates were taken from the Brookhaven Protein Data Bank (PDB).
Their PDB codes are 1AXH, 1AGG, 1EIT, 1VTX, 1OMN, 1OMG and 1GUR respectively.
The structure of the Chinese bird spider toxin Huwentoxin-I (1HWT) was solved
in our laboratory [Qu et al., 1997], which has not been deposited to the
PDB bank. Sequence alignment was performed taking into account that three
disulfide bridges are conserved among all these peptides (Fig. 1).
Table 1 Structure templates used for modeling
|
Code |
Name |
Source |
Activity |
|
1AXH |
Atracotoxin-HVI |
Funnel-web spider toxin |
Insecticidal toxin |
|
1QK6 |
Huwentoxin-I |
Chinese bird spider toxin |
Neuromuscular transmission blocker |
|
1AGG |
Omega-agatoxin-Ivb |
Funnel-web spider toxin |
P-type calcium channel antagonist |
|
1EIT |
mu-agatoxin-I |
Funnel-web spider toxin |
Diverse ion channel specificity |
|
1VTX |
delta-Atracotoxin-HVI |
Funnel-web spider toxin |
Sodium channel blocker |
|
1OMN |
Omega-conotoxin-MVIIc |
Magus cone |
P-type calcium channel antagonist |
|
1OMG |
Omega-conotoxin-MVIIa |
Magus cone |
P-type calcium channel antagonist |
|
1GUR |
Gurmarin |
Gymnema sylvestre |
Sweet taste repressor |
The model building was mainly carried on using the
molecular modeling program Whatif [Vriend, 1998]. The NMR coordinates
of 1AXH were used to build up the backbone fragments. Loops were
searched against the Whatif built-in loop fragment database. The
modeled structure was refined geometrically within Whatif and energy
minimized with the CHARM program to reduce side chain crash.
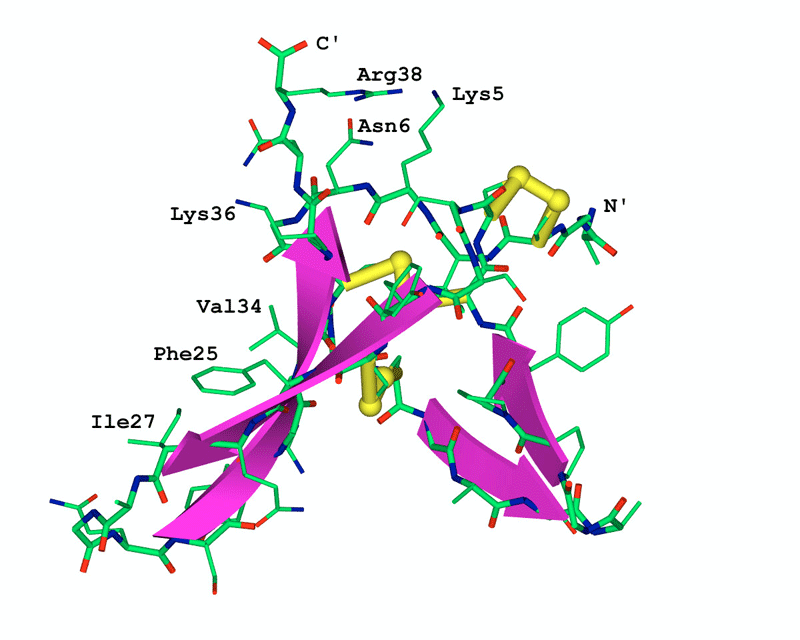
Fig. 2 shows the model of the three dimensional
structure of the anti-fungal peptide. The key feature of this model is the
anti-parallel beta-sheet and the three disulfide bridges, which can be found
in all the 8 templates. The two short strands Fig. 2 can be considered as a
variation among different molecules.
The side chains of three basic residues Lys5, Lys36 and Arg38 located at
one side of the molecule form a positive patch (top in Fig.
2) of the molecule. This implies the possible active site of this anti-fungal
peptide as it was investigated by the mutational analysis that the basic
amino acid residues contribute to the anti-fungal potency [Fant et al.,
1998].
The side chains of three hydrophobic residues Phe25, Ile27 and Val34 sit
at one side of the molecular surface (left side in Fig.
2), which is unusual in molecular packing. Interestingly, this anomalous
hydrophobic surface was also found in the modeling study of the black-eyed
pea trypsin inhibitor which belongs to the cysteine rich Bowman-Birk protease
inhibitor family. The hydrophobic patch along one side of this inhibitor
was explained as a packing force of the possible multimer arrangement of
the protein by both theoretical and experimental study [de Freita et al.,
1997]. Biological experiments are to be carried out on our anti-fungal peptide
to explore understand this structural feature.
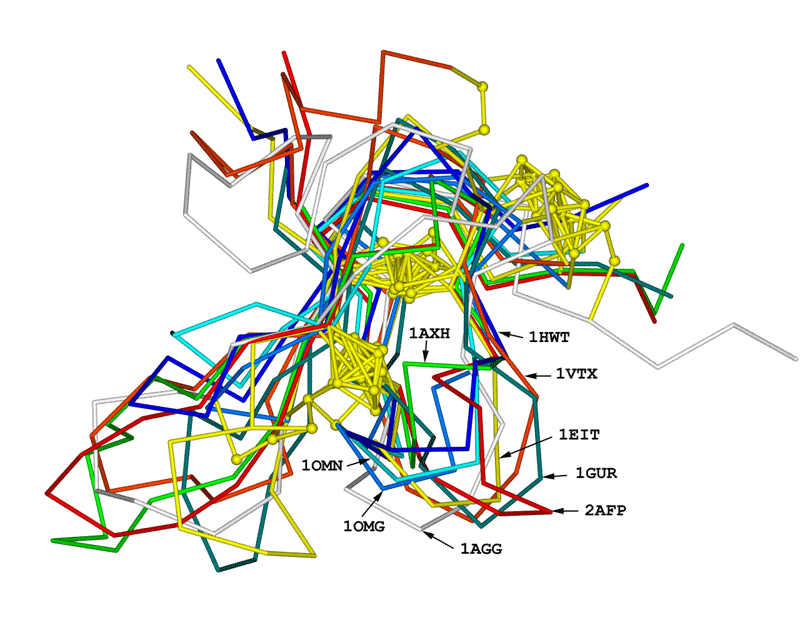
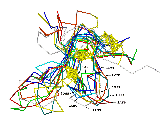
Superimposition of the constructed model onto 8 template shows the structural
similarity of this anti-fungal peptide to all other templates (Fig.
3). The folding unit of these peptides belongs to the cysteine-knot
super family. However, they are different from the anti-fungal peptide from
radish seeds and (1AYJ) and Drosomycin (1MYN) which is featured by the cysteine
stabilized alpha -beta motif. This modeling work, together with the NMR
results from 1AFP, 1AYJ and 1MYN, suggests that different folding units
of anti-fungal peptides may exist, though its evolutional basis is not fully
understood.
AFP: -AGCIKN-GGRCNASAGPPYCCS-SYCFQIAG---QSYGVCKNR
AXH: SPTCIPS-GQPCPYN---ENCCS-QSCTFKENENGNTVKRCD
HWT: --ACKGV-FDACTPG--KNECCPNRVCSDK-------HKWCKWKL
AGG: EDNCIAEDYGKCTWG--GTKCCRGRPCRCSMI---GTNCECTPRLIMEGLSFA
EIT: --ECVPE-NGHCRDW--YDECCEGFYCSCRQ----PPKCICRNNN
VTX: ---CAKK-RNWCGKT---EDCCCPMKCVYAWY---NEQGSCQSTISALWKKC
OMN: ---CKGK-GAPCRKT--MYDCCS-GSCGR--------RGKC
OMG: ---CKGK-GAKCSRL--MYDCCT-GSCRS---------GKC
GUR: --QCVKK-DELCIPY--YLDCCEPLECKKVN----WWDHKCIG
Fig. 1 Sequence alignment of AFP on 8 templates
Six cystein residues which form the three conserved disulfide bridges in all these peptides are in bold face. The paring pattern of the disulfide bridges is
indicated by lines at the top. Dashes denote amino acid residue deletion. The left most code in bold face at each line is the PDB code of the structure
templates for modeling, except for 1HWT (see table 1). 2AFP is the anti-fungal peptide to be modeled. Number of amino acid residues of each
peptide is shown at the right side of each line.
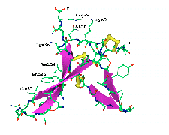
|
Fig. 2 The three-dimensional model of the anti-fungal peptide
The atoms in the constructed three-dimensional model
are presented as sticks with the following color code: green for carbon,
blue for nitrogen, red for oxygen, white for hydrogen and yellow for
sulfide. A gray coil shows the backbone and the beta-strands
are emphasized by red ribbons. The three disulfide bridges are in
thick sticks with the sulfide atoms shown as balls.
|
|
Fig. 3 Structural superimposition of AFP on 8 templates
Structural superimposition of the anti-fungal model
(2AFP) on 8 templates. C-alpha
traces of each peptide are drawn in sticks with different colors.
The side chains of disulfide bridges are shown as ball-stick in yellow.
The PDB code (see table 1) of each chain is indicated with an arrow
line.
|
The pictures of spiders are from Liao-Zhi's thesis. The pictures of pokeberry
are from the Internet.